
DCA Research Team publishes results of Clinical Trials
May 12, 2010
przedruk z:
http://www.dca.med.ualberta.ca/Home/Upd ... Update.cfm
Edmonton, AB - Medical Researchers at the University of Alberta reported today evidence that the orphan generic drug Dichloroacetate (DCA) may hold promise as potential therapy for perhaps the deadliest of all human cancers: a form of brain cancer called glioblastoma. The report is published at the journal Science Translational Medicine, a journal of the American Association of the Advancement of Science; it appears today at the journal's web site http://www.sciencemag.org/
In 2007 the U of A team led by Dr Michelakis, published evidence that DCA reverses cancer growth in non-human models and test tubes. The team showed then that DCA achieves these antitumor effects by altering the metabolism of cancer. By altering the way cancer handles its nutrient fuels, specifically the sugars, DCA was able to take away cancer's most important strength, the resistance to death. Since then, several independent groups across the world have confirmed the Alberta team's findings. In December 2009, the editors of "Science" predicted that cancer metabolism is one of only 5 areas across all scientific disciplines, to "watch for major breakthroughs" in 2010.
The U of A team set out to show that the way that DCA works in actual patients is the same with the way it works in the lab. In addition, researchers wanted to show whether DCA is safe and possibly effective in very sick patients with brain cancer.
By extracting glioblastomas from 49 patients over a period of 2 years and studying them within minutes of removal in the operating room, the team showed that tumors respond to DCA by changing their metabolism. Then, the team treated 5 patients with advanced glioblastoma and secured tumor tissues before and after the DCA therapy. By comparing the two, the team showed that DCA works in these tumors exactly as was predicted by test tube experiments. This is very important because often the results in non-human models tested in the lab do not agree with the results in patients. In addition, the team showed that DCA has anti-cancer effects by altering the metabolism of glioblastoma cancer stem cells, the cells thought responsible for the recurrences of cancer.
In the 5 patients tested, the drug took 3 months to reach blood levels high enough to alter the tumor's metabolism. At those levels, there were no significant adverse effects. However, at some of the higher doses tested, DCA caused nerve malfunction, i.e. numbing of toes and fingers. Importantly, in some patients there was also evidence for clinical benefit, with the tumors either regressing in size or not growing further during the 18 month study.
No conclusions can be made on whether the drug is safe or effective in patients with this form of brain cancer, due to the limited number of patients tested by the study's leads Drs Michelakis and Petruk. Researchers emphasize that use of DCA by patients or physicians, supplied from for-profit sources or without close clinical observation by experienced medical teams in the setting of research trials, is not only inappropriate but may also be dangerous. The U of A results are encouraging and support the need for larger clinical trials with DCA. This work is also one of the first in humans to support the emerging idea that altering the metabolism of tumors is a new direction in the treatment of cancer, Michelakis and Petruk said.
The research team hopes to secure additional funding to continue the ongoing trials with DCA at the University of Alberta. Further studies would include more patients with brain cancer, and test the combination of DCA and standard chemotherapies, eventually including patients from other academic health sciences centres.
One of the intriguing features of this work was that it was funded largely by public donations, including philanthropic foundations and individuals. In addition, it received support by Alberta public institutions, both the University of Alberta and Alberta Health Sciences. The multidisciplinary team that performed this challenging translational research included members of the Departments of Medicine, Diagnostic Imaging and Biomedical Engineering, Oncology and Neurosurgery. Clinicians, scientists, nurses and graduate students worked together for 2 years and express their gratitude to the people of Alberta, philanthropists, the patients and their families.
Dodatkowe info:
http://www.ctv.ca/servlet/ArticleNews/s ... hub=Health
http://www.thedcasite.com/cgi/dcboard.c ... 3&archive=
http://www.calgaryherald.com/health/Cha ... z0onlsiYz3
Wyniki badan klinicznych nad DCA w GBM UA Alberta
Posty: 7
• Strona 1 z 1
Wyniki badan klinicznych nad DCA w GBM UA Alberta
![]() przez Crono5 » Pn maja 24, 2010 1:30 pm
przez Crono5 » Pn maja 24, 2010 1:30 pm
CroNo5-Czarek

http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)

http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
- Crono5
- *** Administrator ***
- Posty: 1424
- Dołączył(a): Wt lut 06, 2007 11:46 pm
- Lokalizacja: Wroclaw /Praszka
Re: Wyniki badan klinicznych nad DCA w GBM UA Alberta
![]() przez Crono5 » Wt maja 25, 2010 12:08 pm
przez Crono5 » Wt maja 25, 2010 12:08 pm
tłumaczenie z:
http://www.dca.med.ualberta.ca/Home/Upd ... Update.cfm
Edmonton, AB – Badacze z University of Alberta donieśli dziś, że mają dowód na to, że sierocy lek Dichlorooctan (DCA/NaDCA) może być potencjalnie obiecującą terapią na najbardziej śmiertelną formę nowotworu spośród wszystkich ludzkich nowotworów – guza mózgu – zwanego glioblastoma. Informacja ta została opublikowana w the journal Science Translational Medicine, czasopiśmie the American Association of the Advancement of Science; dziś (12.05.2010) publikacja ta ukaże się na stronie czasopisma http://www.sciencemag.org/
W 2007 roku zespół badaczy z UA na czele którego stał Dr Michelakis, opublikował dowód, że DCA odwraca wzrost nowotworu w modelach zwierzęcych oraz in vitro. Zespół wykazał, ze DCA osiąga te przeciw nowotworowe właściwości poprzez zmianę metabolizmu nowotworu. Poprzez zmianę sposobu w jaki nowotwór radzi sobie z paliwami odżywczymi, w szczególności cukrami, DCA było wstanie odebrać nowotworowi jego największy atrybut, oporność na śmierć. Od tamtej pory kilka niezależnych grup na świecie potwierdziło doniesienia zespołu z Alberty. W grudniu 2009, redaktorzy Science przewidzieli, że metabolizm jest jedną z tylko 5 wszystkich naukowych dyscyplin w jakiej możemy spodziewać się przełomu w roku 2010.
Zespół badaczy z UA zabrał się za pokazanie, że sposób w jaki DCA działa u pacjentów jest taki sami w jaki robi to w laboratorium. Dodatkowo badacze chcieli wykazać czy DCA jest bezpieczne i możliwie efektywne u bardo chorych pacjentów z nowotworem mózgu.
Poprzez ekstrakcje glioblastoma u 49 pacjentów w okresie 2 lat oraz badając go dosłownie kilka minut po usunięciu w Sali operacyjnej, badacze wykazali, że guzy odpowiadają na DCA poprzez zmianę swojego metabolizmu. Następnie zespół leczył 5 pacjentów z zaawansowaną formą glioblastoma oraz zabezpieczył tkanki guza przed i po terapii z użyciem DCA. Poprzez porównanie obu, zespół wykazał, że DCA działa w przypadku tych guzów, dokładnie w sposób jaki zostało to przewidziane przez badania In vitro. Jest to bardzo istotne ponieważ często wyniki z modeli zwierzęcych nie pokrywają się z wynikami u pacjentów. Dodatkowo zespół wykazał, że poprzez zmianę metabolizmu komórek macierzystych glioblastoma DCA posiada właściwości przeciw nowotworowe. Komórki te odpowiadają za wznowę nowotworu.
U pięciu spośród badanych pacjentów, lekowi zajęło 3 miesiące by był wstanie osiągnąć poziom we krwi na tyle wysoki by mógł zmienić metabolizm guza. Przy tych poziomach nie występowały żadne niepożądane efekty uboczne. Aczkolwiek w przypadku niektórych badanych wyższych dawkach DCA powodował on uszkodzenia nerwów np. drętwienia palców oraz paluchów. Co istotne u niektórych pacjentów obserwowano korzyść kliniczną w postaci albo regresji rozmiaru guza albo braku jego wzrostu w przeciągu 18 miesięcy trwania badania.
W przypadku tej formy nowotworu mózgu z uwagi na ograniczoną ilość pacjentów jaka brała udział w badaniach prowadzących przez Dr Michelakis’a oraz Petruk’a nie można wyciągnąć żadnych wniosków odnośnie efektywności oraz bezpieczeństwa leku wśród pacjentów. Badacze podkreślają, że używanie DCA przez pacjentów lub lekarzy pozyskanego ze źródeł nakierowanych na zysk lub bez dokładnej obserwacji klinicznej przez doświadczone zespoły medyczne w ramach próby klinicznej jest nie tylko nie odpowiednie ale może być niebezpieczne. Wyniki zespołu UA są zachęcające oraz wspierają potrzebę większych badań klinicznych z użyciem DCA. Michelakis oraz Petruk powiedzieli, że ta praca jest jedną z pierwszych prac wśród ludzi która wspiera wschodzący pomysł, że zmiana metabolizmu guzów jest nowym kierunkiem w leczeniu nowotworów.
Zespół badaczy ma nadzieje pozyskać dodatkowe fundusze na kontynuowanie trwającego badania z użyciem DCA mającego miejsce na University of Alberta. W skład dalszych badań wchodziła by większa ilość pacjentów z nowotworem mózgu oraz uwzględniała by przetestowanie połączenia DCA wraz z standardowymi chemioterapiami, ewentualnie uwzględniała by pacjentów z innych akademickich naukowych ośrodków zdrowotnych.
Jednym z intrygujących aspektów tego badania było jego finansowanie w większej części z publicznych datków, wśród których były fundacje filantropijne oraz osoby prywatne. Dodatkowo otrzymało ono wsparcie ze strony instytucji publicznych Alberty, zarówno ze strony University of Alberta jak również Alberta Health Science. W skład multidyscyplinarnego zespołu który podjął się tego translacyjnego badania, wchodzili członkowie wydziałów Medycyny, Obrazowania Diagnostycznego i Inżynierii Biomedycznej, Onkologii oraz Neurochirurgii. Klinicyści, naukowcy, pielęgniarki oraz absolwenci którzy pracowali razem przez okres 2 lat wyrażają wdzięczność ludności Alberty, filantropom oraz pacjentom i ich rodziną.
http://www.dca.med.ualberta.ca/Home/Upd ... Update.cfm
Edmonton, AB – Badacze z University of Alberta donieśli dziś, że mają dowód na to, że sierocy lek Dichlorooctan (DCA/NaDCA) może być potencjalnie obiecującą terapią na najbardziej śmiertelną formę nowotworu spośród wszystkich ludzkich nowotworów – guza mózgu – zwanego glioblastoma. Informacja ta została opublikowana w the journal Science Translational Medicine, czasopiśmie the American Association of the Advancement of Science; dziś (12.05.2010) publikacja ta ukaże się na stronie czasopisma http://www.sciencemag.org/
W 2007 roku zespół badaczy z UA na czele którego stał Dr Michelakis, opublikował dowód, że DCA odwraca wzrost nowotworu w modelach zwierzęcych oraz in vitro. Zespół wykazał, ze DCA osiąga te przeciw nowotworowe właściwości poprzez zmianę metabolizmu nowotworu. Poprzez zmianę sposobu w jaki nowotwór radzi sobie z paliwami odżywczymi, w szczególności cukrami, DCA było wstanie odebrać nowotworowi jego największy atrybut, oporność na śmierć. Od tamtej pory kilka niezależnych grup na świecie potwierdziło doniesienia zespołu z Alberty. W grudniu 2009, redaktorzy Science przewidzieli, że metabolizm jest jedną z tylko 5 wszystkich naukowych dyscyplin w jakiej możemy spodziewać się przełomu w roku 2010.
Zespół badaczy z UA zabrał się za pokazanie, że sposób w jaki DCA działa u pacjentów jest taki sami w jaki robi to w laboratorium. Dodatkowo badacze chcieli wykazać czy DCA jest bezpieczne i możliwie efektywne u bardo chorych pacjentów z nowotworem mózgu.
Poprzez ekstrakcje glioblastoma u 49 pacjentów w okresie 2 lat oraz badając go dosłownie kilka minut po usunięciu w Sali operacyjnej, badacze wykazali, że guzy odpowiadają na DCA poprzez zmianę swojego metabolizmu. Następnie zespół leczył 5 pacjentów z zaawansowaną formą glioblastoma oraz zabezpieczył tkanki guza przed i po terapii z użyciem DCA. Poprzez porównanie obu, zespół wykazał, że DCA działa w przypadku tych guzów, dokładnie w sposób jaki zostało to przewidziane przez badania In vitro. Jest to bardzo istotne ponieważ często wyniki z modeli zwierzęcych nie pokrywają się z wynikami u pacjentów. Dodatkowo zespół wykazał, że poprzez zmianę metabolizmu komórek macierzystych glioblastoma DCA posiada właściwości przeciw nowotworowe. Komórki te odpowiadają za wznowę nowotworu.
U pięciu spośród badanych pacjentów, lekowi zajęło 3 miesiące by był wstanie osiągnąć poziom we krwi na tyle wysoki by mógł zmienić metabolizm guza. Przy tych poziomach nie występowały żadne niepożądane efekty uboczne. Aczkolwiek w przypadku niektórych badanych wyższych dawkach DCA powodował on uszkodzenia nerwów np. drętwienia palców oraz paluchów. Co istotne u niektórych pacjentów obserwowano korzyść kliniczną w postaci albo regresji rozmiaru guza albo braku jego wzrostu w przeciągu 18 miesięcy trwania badania.
W przypadku tej formy nowotworu mózgu z uwagi na ograniczoną ilość pacjentów jaka brała udział w badaniach prowadzących przez Dr Michelakis’a oraz Petruk’a nie można wyciągnąć żadnych wniosków odnośnie efektywności oraz bezpieczeństwa leku wśród pacjentów. Badacze podkreślają, że używanie DCA przez pacjentów lub lekarzy pozyskanego ze źródeł nakierowanych na zysk lub bez dokładnej obserwacji klinicznej przez doświadczone zespoły medyczne w ramach próby klinicznej jest nie tylko nie odpowiednie ale może być niebezpieczne. Wyniki zespołu UA są zachęcające oraz wspierają potrzebę większych badań klinicznych z użyciem DCA. Michelakis oraz Petruk powiedzieli, że ta praca jest jedną z pierwszych prac wśród ludzi która wspiera wschodzący pomysł, że zmiana metabolizmu guzów jest nowym kierunkiem w leczeniu nowotworów.
Zespół badaczy ma nadzieje pozyskać dodatkowe fundusze na kontynuowanie trwającego badania z użyciem DCA mającego miejsce na University of Alberta. W skład dalszych badań wchodziła by większa ilość pacjentów z nowotworem mózgu oraz uwzględniała by przetestowanie połączenia DCA wraz z standardowymi chemioterapiami, ewentualnie uwzględniała by pacjentów z innych akademickich naukowych ośrodków zdrowotnych.
Jednym z intrygujących aspektów tego badania było jego finansowanie w większej części z publicznych datków, wśród których były fundacje filantropijne oraz osoby prywatne. Dodatkowo otrzymało ono wsparcie ze strony instytucji publicznych Alberty, zarówno ze strony University of Alberta jak również Alberta Health Science. W skład multidyscyplinarnego zespołu który podjął się tego translacyjnego badania, wchodzili członkowie wydziałów Medycyny, Obrazowania Diagnostycznego i Inżynierii Biomedycznej, Onkologii oraz Neurochirurgii. Klinicyści, naukowcy, pielęgniarki oraz absolwenci którzy pracowali razem przez okres 2 lat wyrażają wdzięczność ludności Alberty, filantropom oraz pacjentom i ich rodziną.
CroNo5-Czarek
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
- Crono5
- *** Administrator ***
- Posty: 1424
- Dołączył(a): Wt lut 06, 2007 11:46 pm
- Lokalizacja: Wroclaw /Praszka
Re: Wyniki badan klinicznych nad DCA w GBM UA Alberta
![]() przez Crono5 » Wt maja 25, 2010 7:13 pm
przez Crono5 » Wt maja 25, 2010 7:13 pm
Metabolic Modulation of Glioblastoma with Dichloroacetate
przedruk z:
http://stm.sciencemag.org/content/2/31/31ra34.abstract
E. D. Michelakis1,*, G. Sutendra1, P. Dromparis1, L. Webster1, A. Haromy1, E. Niven2, C. Maguire2, T.-L. Gammer1, J. R. Mackey3, D. Fulton3, B. Abdulkarim3, M. S. McMurtry1 and K. C. Petruk4
Solid tumors, including the aggressive primary brain cancer glioblastoma multiforme, develop resistance to cell death, in part as a result of a switch from mitochondrial oxidative phosphorylation to cytoplasmic glycolysis. This metabolic remodeling is accompanied by mitochondrial hyperpolarization. We tested whether the small-molecule and orphan drug dichloroacetate (DCA) can reverse this cancer-specific metabolic and mitochondrial remodeling in glioblastoma. Freshly isolated glioblastomas from 49 patients showed mitochondrial hyperpolarization, which was rapidly reversed by DCA. In a separate experiment with five patients who had glioblastoma, we prospectively secured baseline and serial tumor tissue, developed patient-specific cell lines of glioblastoma and putative glioblastoma stem cells (CD133+, nestin+ cells), and treated each patient with oral DCA for up to 15 months. DCA depolarized mitochondria, increased mitochondrial reactive oxygen species, and induced apoptosis in GBM cells, as well as in putative GBM stem cells, both in vitro and in vivo. DCA therapy also inhibited the hypoxia-inducible factor–1α, promoted p53 activation, and suppressed angiogenesis both in vivo and in vitro. The dose-limiting toxicity was a dose-dependent, reversible peripheral neuropathy, and there was no hematologic, hepatic, renal, or cardiac toxicity. Indications of clinical efficacy were present at a dose that did not cause peripheral neuropathy and at serum concentrations of DCA sufficient to inhibit the target enzyme of DCA, pyruvate dehydrogenase kinase II, which was highly expressed in all glioblastomas. Metabolic modulation may be a viable therapeutic approach in the treatment of glioblastoma.
Citation: E. D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, C. Maguire, T.-L. Gammer, J. R. Mackey, D. Fulton, B. Abdulkarim, M. S. McMurtry, K. C. Petruk, Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2, 31ra34 (2010).
Suplemental data:
http://stm.sciencemag.org/content/suppl ... a34_SM.pdf
przedruk z:
http://stm.sciencemag.org/content/2/31/31ra34.abstract
E. D. Michelakis1,*, G. Sutendra1, P. Dromparis1, L. Webster1, A. Haromy1, E. Niven2, C. Maguire2, T.-L. Gammer1, J. R. Mackey3, D. Fulton3, B. Abdulkarim3, M. S. McMurtry1 and K. C. Petruk4
Solid tumors, including the aggressive primary brain cancer glioblastoma multiforme, develop resistance to cell death, in part as a result of a switch from mitochondrial oxidative phosphorylation to cytoplasmic glycolysis. This metabolic remodeling is accompanied by mitochondrial hyperpolarization. We tested whether the small-molecule and orphan drug dichloroacetate (DCA) can reverse this cancer-specific metabolic and mitochondrial remodeling in glioblastoma. Freshly isolated glioblastomas from 49 patients showed mitochondrial hyperpolarization, which was rapidly reversed by DCA. In a separate experiment with five patients who had glioblastoma, we prospectively secured baseline and serial tumor tissue, developed patient-specific cell lines of glioblastoma and putative glioblastoma stem cells (CD133+, nestin+ cells), and treated each patient with oral DCA for up to 15 months. DCA depolarized mitochondria, increased mitochondrial reactive oxygen species, and induced apoptosis in GBM cells, as well as in putative GBM stem cells, both in vitro and in vivo. DCA therapy also inhibited the hypoxia-inducible factor–1α, promoted p53 activation, and suppressed angiogenesis both in vivo and in vitro. The dose-limiting toxicity was a dose-dependent, reversible peripheral neuropathy, and there was no hematologic, hepatic, renal, or cardiac toxicity. Indications of clinical efficacy were present at a dose that did not cause peripheral neuropathy and at serum concentrations of DCA sufficient to inhibit the target enzyme of DCA, pyruvate dehydrogenase kinase II, which was highly expressed in all glioblastomas. Metabolic modulation may be a viable therapeutic approach in the treatment of glioblastoma.
Citation: E. D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, C. Maguire, T.-L. Gammer, J. R. Mackey, D. Fulton, B. Abdulkarim, M. S. McMurtry, K. C. Petruk, Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2, 31ra34 (2010).
Suplemental data:
http://stm.sciencemag.org/content/suppl ... a34_SM.pdf
CroNo5-Czarek
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
- Crono5
- *** Administrator ***
- Posty: 1424
- Dołączył(a): Wt lut 06, 2007 11:46 pm
- Lokalizacja: Wroclaw /Praszka
Re: Wyniki badan klinicznych nad DCA w GBM UA Alberta
![]() przez Crono5 » Wt maja 25, 2010 8:36 pm
przez Crono5 » Wt maja 25, 2010 8:36 pm
Dodatkowe info
http://scienceblogs.com/insolence/2010/ ... a_vu_a.php
http://scienceblogs.com/insolence/2010/ ... a_vu_a.php
CroNo5-Czarek
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
- Crono5
- *** Administrator ***
- Posty: 1424
- Dołączył(a): Wt lut 06, 2007 11:46 pm
- Lokalizacja: Wroclaw /Praszka
Re: Wyniki badan klinicznych nad DCA w GBM UA Alberta
![]() przez Crono5 » Śr maja 26, 2010 8:32 am
przez Crono5 » Śr maja 26, 2010 8:32 am
Metaboliczna modulacja Glioblastoma z użyciem Dichlorooctanu
Abstrakt
tłumaczenie z:
http://stm.sciencemag.org/content/2/31/31ra34.abstract
E. D. Michelakis1,*, G. Sutendra1, P. Dromparis1, L. Webster1, A. Haromy1, E. Niven2, C. Maguire2, T.-L. Gammer1, J. R. Mackey3, D. Fulton3, B. Abdulkarim3, M. S. McMurtry1 and K. C. Petruk4
Guzy lite, wraz z agresywnym pierwotnym nowotworem mózgu glioblastoma multiforme, wytwarzają oporność na śmierć co jest po części wynikiem przełączenia z mitochondrialnej fosforylacji tlenowej na cytoplazmatyczną glikolizę. Temu metabolicznemu remodelowaniu współtowarzyszy mitochondrialna hiperpolaryzacja. Badaliśmy czy mała molekuła i jednocześnie lek sierocy dichlorooctan (DCA/NaDCA) w przypadku glioblastoma może odwrócić te specyficzne dla nowotworu remodelowanie metaboliczne oraz mitochondrialne. Świeżo wyizolowane glioblastoma spośród 49 pacjentów wykazywało hiperpolaryzację mitochondrialną, która była gwałtownie odwracana przez DCA. W oddzielnym eksperymencie z pięcioma pacjentami którzy mieli glioblastoma, prospektywnie zabezpieczyliśmy podstawową oraz seryjną tkankę guza, która wytworzyła specyficzne dla pacjenta linie komórkowe glioblastoma oraz domniemane komórki macierzyste glioblastoma (CD133+, nestin+ komórki), po czym następnie leczyliśmy każdego pacjenta z użyciem oralnej dawki DCA(NaDCA) przez okres do 15 miesięcy. DCA zarówno In vitro jak i In vivo depolaryzowało mitochondria, zwiększało poziom mitochondrialnych reaktywnych form tlenu (ROS) oraz indukowało apoptozę w komórkach GBM, jak również w domniemanych komórkach macierzystych GBM. Terapia z użyciem DCA zarówno In vivo jak In vitro inhibitowała czynnik indukowany hipoksją typu 1a, promowała aktywacje p53 oraz tłumiła angiogiogenezę. Toksyczność ograniczona dawką była zależna od dawki, występowała odwracalna neuropatia obwodowa brak było natomiast hematologicznych, wątrobowych, nerkowych oraz kardiologicznych toksyczności. Wskazania klinicznej efektywności były obecne w obrębie dawki która nie powodowała neuropatii obwodowej oraz przy koncentracji DCA w serum krwi wystarczającej do tłumienia docelowego enzymu DCA, kinazy II dehydrogenazy pirogronianowej, która była silnie ekpspresjonowana we wszystkich glioblastoma. Modulacja metaboliczna może być wartościowym podejściem terapeutycznym w leczeniu glioblastoma.
dane uzupełniające:
http://stm.sciencemag.org/content/suppl ... a34_SM.pdf
dodatkowe info:
http://scienceblogs.com/insolence/2010/ ... a_vu_a.php
Abstrakt
tłumaczenie z:
http://stm.sciencemag.org/content/2/31/31ra34.abstract
E. D. Michelakis1,*, G. Sutendra1, P. Dromparis1, L. Webster1, A. Haromy1, E. Niven2, C. Maguire2, T.-L. Gammer1, J. R. Mackey3, D. Fulton3, B. Abdulkarim3, M. S. McMurtry1 and K. C. Petruk4
Guzy lite, wraz z agresywnym pierwotnym nowotworem mózgu glioblastoma multiforme, wytwarzają oporność na śmierć co jest po części wynikiem przełączenia z mitochondrialnej fosforylacji tlenowej na cytoplazmatyczną glikolizę. Temu metabolicznemu remodelowaniu współtowarzyszy mitochondrialna hiperpolaryzacja. Badaliśmy czy mała molekuła i jednocześnie lek sierocy dichlorooctan (DCA/NaDCA) w przypadku glioblastoma może odwrócić te specyficzne dla nowotworu remodelowanie metaboliczne oraz mitochondrialne. Świeżo wyizolowane glioblastoma spośród 49 pacjentów wykazywało hiperpolaryzację mitochondrialną, która była gwałtownie odwracana przez DCA. W oddzielnym eksperymencie z pięcioma pacjentami którzy mieli glioblastoma, prospektywnie zabezpieczyliśmy podstawową oraz seryjną tkankę guza, która wytworzyła specyficzne dla pacjenta linie komórkowe glioblastoma oraz domniemane komórki macierzyste glioblastoma (CD133+, nestin+ komórki), po czym następnie leczyliśmy każdego pacjenta z użyciem oralnej dawki DCA(NaDCA) przez okres do 15 miesięcy. DCA zarówno In vitro jak i In vivo depolaryzowało mitochondria, zwiększało poziom mitochondrialnych reaktywnych form tlenu (ROS) oraz indukowało apoptozę w komórkach GBM, jak również w domniemanych komórkach macierzystych GBM. Terapia z użyciem DCA zarówno In vivo jak In vitro inhibitowała czynnik indukowany hipoksją typu 1a, promowała aktywacje p53 oraz tłumiła angiogiogenezę. Toksyczność ograniczona dawką była zależna od dawki, występowała odwracalna neuropatia obwodowa brak było natomiast hematologicznych, wątrobowych, nerkowych oraz kardiologicznych toksyczności. Wskazania klinicznej efektywności były obecne w obrębie dawki która nie powodowała neuropatii obwodowej oraz przy koncentracji DCA w serum krwi wystarczającej do tłumienia docelowego enzymu DCA, kinazy II dehydrogenazy pirogronianowej, która była silnie ekpspresjonowana we wszystkich glioblastoma. Modulacja metaboliczna może być wartościowym podejściem terapeutycznym w leczeniu glioblastoma.
dane uzupełniające:
http://stm.sciencemag.org/content/suppl ... a34_SM.pdf
dodatkowe info:
http://scienceblogs.com/insolence/2010/ ... a_vu_a.php
CroNo5-Czarek
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
- Crono5
- *** Administrator ***
- Posty: 1424
- Dołączył(a): Wt lut 06, 2007 11:46 pm
- Lokalizacja: Wroclaw /Praszka
Re: Wyniki badan klinicznych nad DCA w GBM UA Alberta
![]() przez Crono5 » N maja 30, 2010 11:26 am
przez Crono5 » N maja 30, 2010 11:26 am
Dane uzupełniające
tłumaczenie fragmentów z:
http://stm.sciencemag.org/content/suppl ... a34_SM.pdf
Przypadki kliniczne
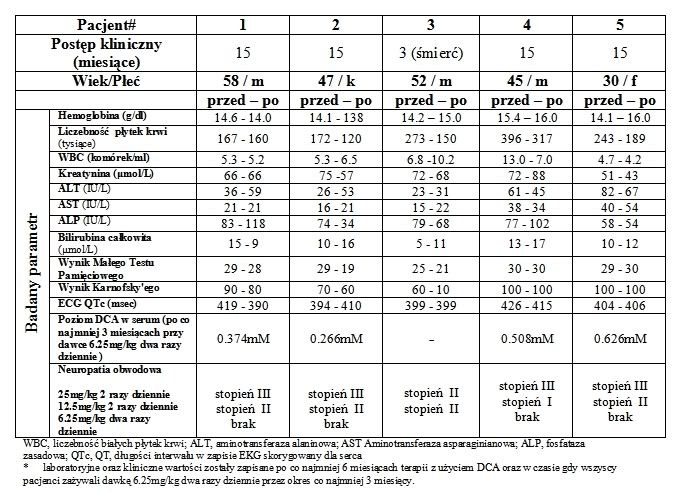
Pacjent 1 jest 58 letnim mężczyznom u którego nastąpiła wznowa GBM 4 miesiące po zakończeniu standardowej terapii na którą składał się wstępny zabiegiem usunięcia masy guza, radioterapia, oraz TMZ (Temodal). W tym okresie (14 miesięcy po resekcji) został skierowany na terapię DCA. Jego sprawność wg. skali Karnofsky'ego (KPS) wynosiła 90 punktów, nie doświadczał także znaczącego obrzęku mózgu. Po piętnastu miesiącach trwania terapii z użyciem DCA (dawka 6.25mg/kg podawana doustnie dwa razy dziennie od miesiąca 7) jego stan kliniczny pozostawał stabilny pierwotna lokalizacja guza również pozostawała stabilna i nie ulegała wzrostowi. Ponadto występowała znacząca regresja w dwóch wtórnych masach przy-skroniowych (Ryc. 2A i S2).
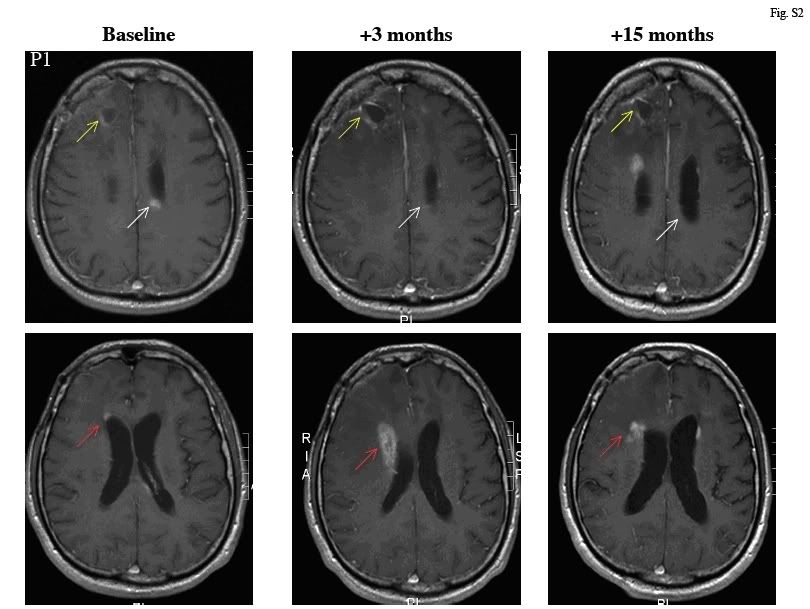
Ryc. S2 Rozwój odpowiedzi guza u pacjenta 1[/b]
Pacjent 2 to 47 letnia kobieta z historią kilku wznów GBM pomimo zastosowania resekcji. Radioterapii, TMZ oraz kilku protokołów chemioterapii paliatywnej, włączając Etopozyd, CCNU (Lomustyna) oraz Tamoksyfen. Jej ostatnia chemioterapia została zakończona cztery miesiące przed zwerbowaniem. Cierpiała z powodu prawostronnego zaburzenia ruchowo – czuciowego oraz, aż do jej ostatniej wznowy, miała przeprowadzony zabieg resekcji, po którym nastąpiła terapia z użyciem DCA. Drugi zabieg resekcji oraz drenaż torbieli objawowych został przeprowadzony 11 miesiąca. Piętnaście miesięcy po rozpoczęciu terapii z użyciem DCA, jej stan kliniczny pozostawał stabilny z utrzymującą się poprawą radiologiczną (Ryc. 2A)
Pacjentem 3 był 52 letni mężczyzna z historią kilku wznów GBM pomimo zabiegu resekcji, radioterapii, TMZ oraz kilku protokołów terapii paliatywnej, w skład których wchodziły Etopozyd, CCNU (Lomustyna) oraz Tamoksyfen. Ostatni zabieg resekcji został u niego przeprowadzony 3 miesiące przed zwerbowaniem. Od czasu jego ostatniej progresji schorzenia był nieoperacyjny z uwagi na duży rozmiar guza oraz znaczący obrzęk z przesunięciem linii środkowej pomimo podawania maksymalnych dawek sterydów (Ryc.S5)
Podjął się terapii z użyciem DCA, a jego poziom KPS w tym okresie wynosił 60 punktów. Postępujące nadciśnienie śródczaszkowe doprowadziło do jego śmierci 3 miesiące później.
Ryc. S5 GBM RM pacjenta 3
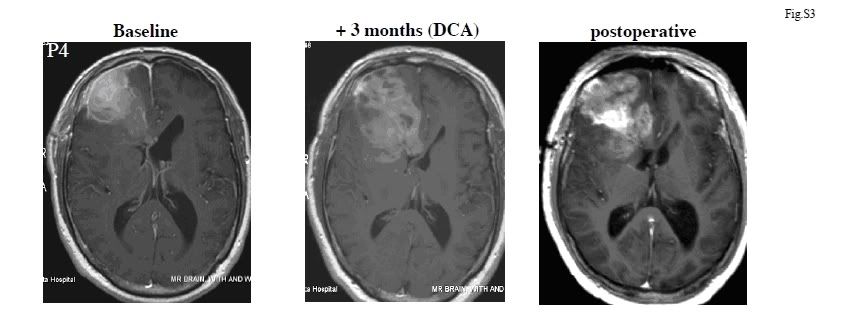
Pacjent 4 to 45 letni mężczyzna który przeszedł wstępną resekcję po czym wszedł do 3 miesięcznego protokołu przed terapii z użyciem DCA po czym nastąpiło podawanie DCA + standardowa terapia. Było to oparte na spekulacji, że DCA może uczulić guz na dalszą chemioterapię. Aczkolwiek pod koniec trzeciego miesiąca wykazywał radiologiczny dowód na progresje schorzenia, więc został przeprowadzony drugi zabieg resekcji. Po zabiegu kontynuował on zażywanie DCA wraz z standardową terapią (radioterapia oraz TMZ) Jego terapia z użyciem TMZ została wydłużona do 9 miesięcy, w oparciu o standardową praktykę jaka była w naszym programie, ponieważ wykazywał nieprzerwaną regresję. Po fazie kombinowanej kontynuował zażywanie samego DCA przez okres 6 miesięcy. W tym okresie (18 miesięcy terapii z użyciem DCA, 15 miesięcy po tym jak terapia kombinowana została rozpoczęta) pacjent pozostawał bezobjawowy z KPS na poziomie 100 punktów, oraz brakiem radiologicznych oznak na wzrost guza lub wznowę. (Ryc. S3)
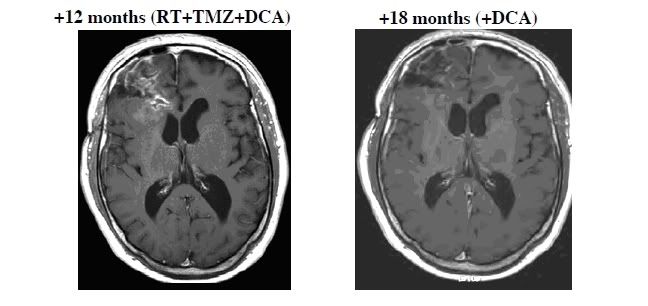
Ryc. S3 Rozwój odpowiedzi guza u pacjenta 4
Pacjentem 5 jest 30 letnia kobieta po zabiegu resekcji GBM, po którym rozpoczęła terapię z użyciem DCA wraz z radioterapią i terapią z użyciem TMZ. Sześć miesięcy później ukończyła 6 miesięczny reżim TMZ i zażywała nadal samo DCA przed dodatkowe 9 miesięcy. Był dowód na kontynuacje regresji po tym jak terapia z użyciem TMZ została zakończona oraz 15 miesięcy po zwerbowaniu (15 miesięcy zażywania DCA) wykazywała całkowite ustąpienie guza (Ryc. S4), pozostając bezobjawowa z KPS na poziomie 100 punktów.
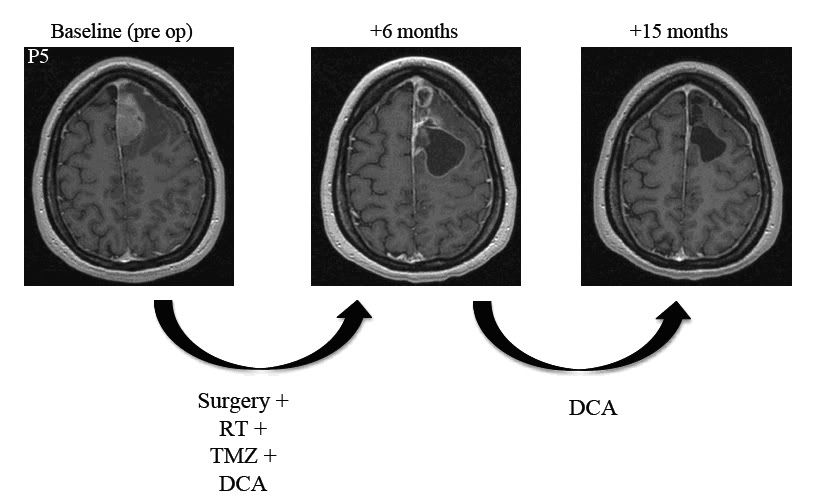
Ryc. S4 Rozwój odpowiedzi guza u pacjenta 5.
Dyskusja
Proponowany ogólny mechanizm przeciw nowotworowych właściwości DCA w przypadku GBM (Ryc. S12)
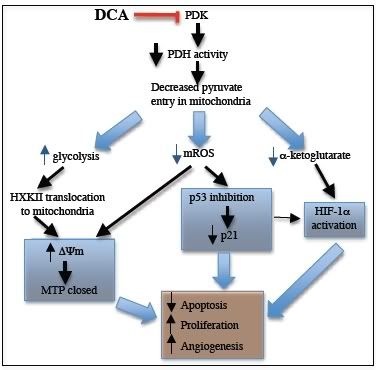
Ryc. S12 Proponowany ogólny mechanizm przeciw nowotworowych właściwości DCA w przypadku GBM
Poprzednio wykazaliśmy, ze DCA indukuje apoptozę zależną od mitochondriów w kilku liniach komórkowych, poprzez inhibitowanie mitochondrialnych enzymó kinazy dehydrogenazy pirogronianowej (PDK) (1).
Wykazaliśmy, że skutkuje to wzrostem współczynnika oksydacji glukozy i jest związane z depolaryzacją mitochondriów oraz wzrostem produkcji mROS w kilku liniach nowotworów(1).
Obecnie wykazujemy, że DCA indukuje apoptozę, depolaryzuje mitochondria oraz zwiększa poziom mROS w ludzkich guzach GBM (Ryc. 1, 2B,5C), pierwotnych liniach komórkowych GBM (Ryc. 3B, S9) oraz rzekomych GBM-S.C. (Ryc. 3B, 4, 5C, S9). Indukowana przez DCA aktywacja PDH (Ryc. 2C) promuje oksydatywną dekarboksylacja pirogronianu do acetyl-CoA, głownego substratu cyklu Krebsa w macierzy mitochondrialnej. W wyniku tego DCA zwiększa koncentracje produkty cyklu Krebsa kwasu alfa-ketoglutarowego (Ryc. 6) jak również donorów elektronowych (NADH, FADH2)(1) które są dawcami elektronów w łańcuchu transportu elektronów zwiększając oddychanie (o ~40% zobacz wyniki) oraz produkcję mROS. Wzrost produkcji kwasu alfa-ketoglutarowego oraz ponadtlenków mają dodatkowo istotny dalszy wpływ, ponieważ mogą one zdestabilizować HIF-1α oraz aktywować p53, oba które były związane z patogenezą GBM.
Na przykład kwas alfa-ketoglutarowy jest krytycznym współczynnikiem prolyl-hydroxylases który destabilizuje HIF-1α (2). Dodatkowo obecność manganowej dysmutazy ponadtlenkowej (MnSOD), ponadtlenek jest dyskutowany do H2O2 który jest bardziej stabilny i może osiągnąć dodatkowe cele mitochondrialne, takie jak błonowe kanały potasowe (1) albo czynniki redox wyczulone transkrypcyjne (3 – 5). DCA zwiększa poziomy H2O2 które w połączeniu z zwiększonymi poziomami kwasu alfa-ketoglutarowego (Ryc. 6) mogą wyjaśniać inhibicję HIF-1α. Wykazaliśmy, że HIF-1α. Jest inhibitowany w guzach tkanek GBM pochodzących od pacjentów, w porównaniu z tkankami od tych samych pacjentów z sprzed terapii z użyciem DCA (Ryc. 5B). Utrzymując spadek aktywności HIF-1α, DCA zmniejsza także koncentracje VEGF w komórkach GBM (Ryc. 6) co może tłumaczyć obniżony poziom angiogenezy (odwrotnie do tego co przewidywał Clooney (viewtopic.php?p=9390#p9390) –dop. corn) co wykazujemy w tkankach GBM pochodzących od chronicznie leczonych pacjentów (Ryc. 5A, S9D) oraz jak również In vitro (Ryc. S19). Podwyższenie poziomu nadtlenku oraz H2O2 może tłumaczyć aktywacje czułego na redox p53 (przeniesienie jądrowe) (5) które to wykazaliśmy w tkankach GBM pochodzących od chronicznie leczonych pacjentów (Ryc. S11). W tych samych komórkach p53 jest aktywowane, jego dalszy cel p21 jest zarówno regulowany w górę oraz aktywowany (przeniesienie jądrowe) (Ryc. S11) Aktywacja p21 może również wyjaśniać zmniejszone współczynniki proliferacji które wykazaliśmy w przypadku komórek GBM zarówno In vitro (Ryc. S9C) oraz w tkankach GBM po chronicznej terapii z użyciem DCA (Ryc.2B). Aktywacja p53 może również przyczyniać się do inhibicji HIF-1α, ponieważ oba czynniki transkrypcji współzawodniczą o ten sam kofaktor (6 - 8). Główny spadek w aktywności PDH oraz współczynników oddechowych w GBM (nawet w trakcie normoxia ) mogą indukować mitochodrialną „pseudo hipoksyczne” sygnały które mogą się przyczyniać do aktywacji HIF-1α oraz tłumienia p53. Aktywacja PDH przez DCA może eliminować te pseudo hipoksyczne sygnały i przyczyniać się do obserwowanych anty angiogennych właściwości które są terapeutycznie istotne w tych bardzo unaczynionych guzach.
Ryc. S9 Wpływ terapii z użyciem DCA na GBM – S.C. oraz apoptozę naczyniową
Poprzednio wykazaliśmy, że DCA może depolaryzować mitochondria po części poprzez otwieranie VDAC, ponieważ skutkom jego działania częściowo zapobiegały inhibitory VDAC (1). Pastorino et al, wykazał, że potencjalny mechanizm nowotworowej hiperpolaryzacji mitochondriów [co charakteryzuję większość guzów litych (9)] jest translokacją glikolitycznego oraz uwrażliwionego na HIF enzymu Heksokinazy II (HXK-II) z cytoplazmy do zewnętrznej błony mitochondrialnej gdzie wiąże i tłumi VDAC (10, 11). Obecnie wykazaliśmy że pierwotne linie komórkowe GBM pochodzące od pacjentów sprzed terapii z użyciem DCA posiadały HXK-II zlokalizowane z mitochondriach (Ryc. S8) potwierdzając spostrzeżenie Pastorino. Aczkolwiek w pierwotnych liniach komórkowych GBM pochodzących z guzów po terapii z użyciem DCA uległo to normalizacji i HXK-II nie był już zlokalizowany z mitochondriami, zgodnie z depolaryzacją mitochondriów indokwaną przez DCA.
Ryc. S8 Heksokinaza II (HXK-II) w komórkach GBM pozyskanych od pacjentów przed i po chronicznej terapii z użyciem DCA
Jednakże dokładna rola VDAC oraz MTP w nowotworach oraz apoptozie pozostaje niejasna (12) . Dodatkowo mechanizm przez który DCA zwiększa poziom mROS, w czasie kiedy depolaryzuje mitochondria pozostaje niejasny. Poprzednio wykazaliśmy, że źródłem mROS w komórkach leczonych z użyciem DCA jest Łańcuch transportu elektronów kompleks I (1). Zaproponowaliśmy mechanizm za pośrednictwem którego kompleks I (największy ze wszystkich kompleksów transportu elektronów oraz najbardziej podatny na uszkodzenia oksydacyjne z uwagi na jego duża zawartość grup żelazowo – siarkowych ) jest inhibitowany przez zwiększenie poziomu mROS indukowane przez DCA (1).
Może to wyjaśniać ewentualny spadek potencjału błony mitochondrialnej oraz inicjację apoptozy w sposób podobny do tego w liniach komórkowych w których kompleks I jest genetycznie inhibitowany, które charakteryzują się zarówno zwiększonym poziomem mROS oraz depolaryzacją mitochondriów (13,14)
In vivo DCA może mieć dodatkowe właściwości metaboliczne, wykraczające poza te obserwowane w kontrolowanych warunkach metabolizmu kultury. Na przykład pirogronian może wejść w ważne reakcje anaplerotyczne oraz ścieżki biosyntezy aminokwasów, które zostały zaproponowane jako ważne dla przeżycia komórki nowotworowej (15).
Co więcej aktywacja PDH oraz oksydacji glukozy może pośrednio [poprzez Cykl Randla (16)] inhibitować oksydacje kwasów tłuszczowych, co również zostało wykazane, że jest istotne dla przeżycia komórki nowotworowej (15).
Ryc. S10 Wpływ DCA na angiogenezę In vivo
Ryc.S11 Wpływ terapii z użyciem DCA na aktywność In vivo p53 oraz p21
Referencje
tłumaczenie fragmentów z:
http://stm.sciencemag.org/content/suppl ... a34_SM.pdf
Przypadki kliniczne
Pacjent 1 jest 58 letnim mężczyznom u którego nastąpiła wznowa GBM 4 miesiące po zakończeniu standardowej terapii na którą składał się wstępny zabiegiem usunięcia masy guza, radioterapia, oraz TMZ (Temodal). W tym okresie (14 miesięcy po resekcji) został skierowany na terapię DCA. Jego sprawność wg. skali Karnofsky'ego (KPS) wynosiła 90 punktów, nie doświadczał także znaczącego obrzęku mózgu. Po piętnastu miesiącach trwania terapii z użyciem DCA (dawka 6.25mg/kg podawana doustnie dwa razy dziennie od miesiąca 7) jego stan kliniczny pozostawał stabilny pierwotna lokalizacja guza również pozostawała stabilna i nie ulegała wzrostowi. Ponadto występowała znacząca regresja w dwóch wtórnych masach przy-skroniowych (Ryc. 2A i S2).
Ryc. S2 Rozwój odpowiedzi guza u pacjenta 1[/b]
Pacjent 2 to 47 letnia kobieta z historią kilku wznów GBM pomimo zastosowania resekcji. Radioterapii, TMZ oraz kilku protokołów chemioterapii paliatywnej, włączając Etopozyd, CCNU (Lomustyna) oraz Tamoksyfen. Jej ostatnia chemioterapia została zakończona cztery miesiące przed zwerbowaniem. Cierpiała z powodu prawostronnego zaburzenia ruchowo – czuciowego oraz, aż do jej ostatniej wznowy, miała przeprowadzony zabieg resekcji, po którym nastąpiła terapia z użyciem DCA. Drugi zabieg resekcji oraz drenaż torbieli objawowych został przeprowadzony 11 miesiąca. Piętnaście miesięcy po rozpoczęciu terapii z użyciem DCA, jej stan kliniczny pozostawał stabilny z utrzymującą się poprawą radiologiczną (Ryc. 2A)
Pacjentem 3 był 52 letni mężczyzna z historią kilku wznów GBM pomimo zabiegu resekcji, radioterapii, TMZ oraz kilku protokołów terapii paliatywnej, w skład których wchodziły Etopozyd, CCNU (Lomustyna) oraz Tamoksyfen. Ostatni zabieg resekcji został u niego przeprowadzony 3 miesiące przed zwerbowaniem. Od czasu jego ostatniej progresji schorzenia był nieoperacyjny z uwagi na duży rozmiar guza oraz znaczący obrzęk z przesunięciem linii środkowej pomimo podawania maksymalnych dawek sterydów (Ryc.S5)
Podjął się terapii z użyciem DCA, a jego poziom KPS w tym okresie wynosił 60 punktów. Postępujące nadciśnienie śródczaszkowe doprowadziło do jego śmierci 3 miesiące później.
Ryc. S5 GBM RM pacjenta 3
Pacjent 4 to 45 letni mężczyzna który przeszedł wstępną resekcję po czym wszedł do 3 miesięcznego protokołu przed terapii z użyciem DCA po czym nastąpiło podawanie DCA + standardowa terapia. Było to oparte na spekulacji, że DCA może uczulić guz na dalszą chemioterapię. Aczkolwiek pod koniec trzeciego miesiąca wykazywał radiologiczny dowód na progresje schorzenia, więc został przeprowadzony drugi zabieg resekcji. Po zabiegu kontynuował on zażywanie DCA wraz z standardową terapią (radioterapia oraz TMZ) Jego terapia z użyciem TMZ została wydłużona do 9 miesięcy, w oparciu o standardową praktykę jaka była w naszym programie, ponieważ wykazywał nieprzerwaną regresję. Po fazie kombinowanej kontynuował zażywanie samego DCA przez okres 6 miesięcy. W tym okresie (18 miesięcy terapii z użyciem DCA, 15 miesięcy po tym jak terapia kombinowana została rozpoczęta) pacjent pozostawał bezobjawowy z KPS na poziomie 100 punktów, oraz brakiem radiologicznych oznak na wzrost guza lub wznowę. (Ryc. S3)
Ryc. S3 Rozwój odpowiedzi guza u pacjenta 4
Pacjentem 5 jest 30 letnia kobieta po zabiegu resekcji GBM, po którym rozpoczęła terapię z użyciem DCA wraz z radioterapią i terapią z użyciem TMZ. Sześć miesięcy później ukończyła 6 miesięczny reżim TMZ i zażywała nadal samo DCA przed dodatkowe 9 miesięcy. Był dowód na kontynuacje regresji po tym jak terapia z użyciem TMZ została zakończona oraz 15 miesięcy po zwerbowaniu (15 miesięcy zażywania DCA) wykazywała całkowite ustąpienie guza (Ryc. S4), pozostając bezobjawowa z KPS na poziomie 100 punktów.
Ryc. S4 Rozwój odpowiedzi guza u pacjenta 5.
Dyskusja
Proponowany ogólny mechanizm przeciw nowotworowych właściwości DCA w przypadku GBM (Ryc. S12)
Ryc. S12 Proponowany ogólny mechanizm przeciw nowotworowych właściwości DCA w przypadku GBM
Poprzednio wykazaliśmy, ze DCA indukuje apoptozę zależną od mitochondriów w kilku liniach komórkowych, poprzez inhibitowanie mitochondrialnych enzymó kinazy dehydrogenazy pirogronianowej (PDK) (1).
Wykazaliśmy, że skutkuje to wzrostem współczynnika oksydacji glukozy i jest związane z depolaryzacją mitochondriów oraz wzrostem produkcji mROS w kilku liniach nowotworów(1).
Obecnie wykazujemy, że DCA indukuje apoptozę, depolaryzuje mitochondria oraz zwiększa poziom mROS w ludzkich guzach GBM (Ryc. 1, 2B,5C), pierwotnych liniach komórkowych GBM (Ryc. 3B, S9) oraz rzekomych GBM-S.C. (Ryc. 3B, 4, 5C, S9). Indukowana przez DCA aktywacja PDH (Ryc. 2C) promuje oksydatywną dekarboksylacja pirogronianu do acetyl-CoA, głownego substratu cyklu Krebsa w macierzy mitochondrialnej. W wyniku tego DCA zwiększa koncentracje produkty cyklu Krebsa kwasu alfa-ketoglutarowego (Ryc. 6) jak również donorów elektronowych (NADH, FADH2)(1) które są dawcami elektronów w łańcuchu transportu elektronów zwiększając oddychanie (o ~40% zobacz wyniki) oraz produkcję mROS. Wzrost produkcji kwasu alfa-ketoglutarowego oraz ponadtlenków mają dodatkowo istotny dalszy wpływ, ponieważ mogą one zdestabilizować HIF-1α oraz aktywować p53, oba które były związane z patogenezą GBM.
Na przykład kwas alfa-ketoglutarowy jest krytycznym współczynnikiem prolyl-hydroxylases który destabilizuje HIF-1α (2). Dodatkowo obecność manganowej dysmutazy ponadtlenkowej (MnSOD), ponadtlenek jest dyskutowany do H2O2 który jest bardziej stabilny i może osiągnąć dodatkowe cele mitochondrialne, takie jak błonowe kanały potasowe (1) albo czynniki redox wyczulone transkrypcyjne (3 – 5). DCA zwiększa poziomy H2O2 które w połączeniu z zwiększonymi poziomami kwasu alfa-ketoglutarowego (Ryc. 6) mogą wyjaśniać inhibicję HIF-1α. Wykazaliśmy, że HIF-1α. Jest inhibitowany w guzach tkanek GBM pochodzących od pacjentów, w porównaniu z tkankami od tych samych pacjentów z sprzed terapii z użyciem DCA (Ryc. 5B). Utrzymując spadek aktywności HIF-1α, DCA zmniejsza także koncentracje VEGF w komórkach GBM (Ryc. 6) co może tłumaczyć obniżony poziom angiogenezy (odwrotnie do tego co przewidywał Clooney (viewtopic.php?p=9390#p9390) –dop. corn) co wykazujemy w tkankach GBM pochodzących od chronicznie leczonych pacjentów (Ryc. 5A, S9D) oraz jak również In vitro (Ryc. S19). Podwyższenie poziomu nadtlenku oraz H2O2 może tłumaczyć aktywacje czułego na redox p53 (przeniesienie jądrowe) (5) które to wykazaliśmy w tkankach GBM pochodzących od chronicznie leczonych pacjentów (Ryc. S11). W tych samych komórkach p53 jest aktywowane, jego dalszy cel p21 jest zarówno regulowany w górę oraz aktywowany (przeniesienie jądrowe) (Ryc. S11) Aktywacja p21 może również wyjaśniać zmniejszone współczynniki proliferacji które wykazaliśmy w przypadku komórek GBM zarówno In vitro (Ryc. S9C) oraz w tkankach GBM po chronicznej terapii z użyciem DCA (Ryc.2B). Aktywacja p53 może również przyczyniać się do inhibicji HIF-1α, ponieważ oba czynniki transkrypcji współzawodniczą o ten sam kofaktor (6 - 8). Główny spadek w aktywności PDH oraz współczynników oddechowych w GBM (nawet w trakcie normoxia ) mogą indukować mitochodrialną „pseudo hipoksyczne” sygnały które mogą się przyczyniać do aktywacji HIF-1α oraz tłumienia p53. Aktywacja PDH przez DCA może eliminować te pseudo hipoksyczne sygnały i przyczyniać się do obserwowanych anty angiogennych właściwości które są terapeutycznie istotne w tych bardzo unaczynionych guzach.
Ryc. S9 Wpływ terapii z użyciem DCA na GBM – S.C. oraz apoptozę naczyniową
Poprzednio wykazaliśmy, że DCA może depolaryzować mitochondria po części poprzez otwieranie VDAC, ponieważ skutkom jego działania częściowo zapobiegały inhibitory VDAC (1). Pastorino et al, wykazał, że potencjalny mechanizm nowotworowej hiperpolaryzacji mitochondriów [co charakteryzuję większość guzów litych (9)] jest translokacją glikolitycznego oraz uwrażliwionego na HIF enzymu Heksokinazy II (HXK-II) z cytoplazmy do zewnętrznej błony mitochondrialnej gdzie wiąże i tłumi VDAC (10, 11). Obecnie wykazaliśmy że pierwotne linie komórkowe GBM pochodzące od pacjentów sprzed terapii z użyciem DCA posiadały HXK-II zlokalizowane z mitochondriach (Ryc. S8) potwierdzając spostrzeżenie Pastorino. Aczkolwiek w pierwotnych liniach komórkowych GBM pochodzących z guzów po terapii z użyciem DCA uległo to normalizacji i HXK-II nie był już zlokalizowany z mitochondriami, zgodnie z depolaryzacją mitochondriów indokwaną przez DCA.
Ryc. S8 Heksokinaza II (HXK-II) w komórkach GBM pozyskanych od pacjentów przed i po chronicznej terapii z użyciem DCA
Jednakże dokładna rola VDAC oraz MTP w nowotworach oraz apoptozie pozostaje niejasna (12) . Dodatkowo mechanizm przez który DCA zwiększa poziom mROS, w czasie kiedy depolaryzuje mitochondria pozostaje niejasny. Poprzednio wykazaliśmy, że źródłem mROS w komórkach leczonych z użyciem DCA jest Łańcuch transportu elektronów kompleks I (1). Zaproponowaliśmy mechanizm za pośrednictwem którego kompleks I (największy ze wszystkich kompleksów transportu elektronów oraz najbardziej podatny na uszkodzenia oksydacyjne z uwagi na jego duża zawartość grup żelazowo – siarkowych ) jest inhibitowany przez zwiększenie poziomu mROS indukowane przez DCA (1).
Może to wyjaśniać ewentualny spadek potencjału błony mitochondrialnej oraz inicjację apoptozy w sposób podobny do tego w liniach komórkowych w których kompleks I jest genetycznie inhibitowany, które charakteryzują się zarówno zwiększonym poziomem mROS oraz depolaryzacją mitochondriów (13,14)
In vivo DCA może mieć dodatkowe właściwości metaboliczne, wykraczające poza te obserwowane w kontrolowanych warunkach metabolizmu kultury. Na przykład pirogronian może wejść w ważne reakcje anaplerotyczne oraz ścieżki biosyntezy aminokwasów, które zostały zaproponowane jako ważne dla przeżycia komórki nowotworowej (15).
Co więcej aktywacja PDH oraz oksydacji glukozy może pośrednio [poprzez Cykl Randla (16)] inhibitować oksydacje kwasów tłuszczowych, co również zostało wykazane, że jest istotne dla przeżycia komórki nowotworowej (15).
Ryc. S10 Wpływ DCA na angiogenezę In vivo
Ryc.S11 Wpływ terapii z użyciem DCA na aktywność In vivo p53 oraz p21
Referencje
CroNo5-Czarek
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
- Crono5
- *** Administrator ***
- Posty: 1424
- Dołączył(a): Wt lut 06, 2007 11:46 pm
- Lokalizacja: Wroclaw /Praszka
Re: Wyniki badan klinicznych nad DCA w GBM UA Alberta
![]() przez Crono5 » So cze 19, 2010 1:17 am
przez Crono5 » So cze 19, 2010 1:17 am
CroNo5-Czarek
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
http://dca-information.pbworks.com/
http://apoptoza.info
http://thedcasite.com
http://www.curcuminresearch.org/cancer.html
http://www.vitamindwiki.com/VitaminDWiki
http://science-news.blog.onet.pl
Contra spem, spero (wbrew wszystkim, wierzę)
- Crono5
- *** Administrator ***
- Posty: 1424
- Dołączył(a): Wt lut 06, 2007 11:46 pm
- Lokalizacja: Wroclaw /Praszka
Posty: 7
• Strona 1 z 1
Kto przegląda forum
Użytkownicy przeglądający ten dział: Brak zidentyfikowanych użytkowników i 6 gości